
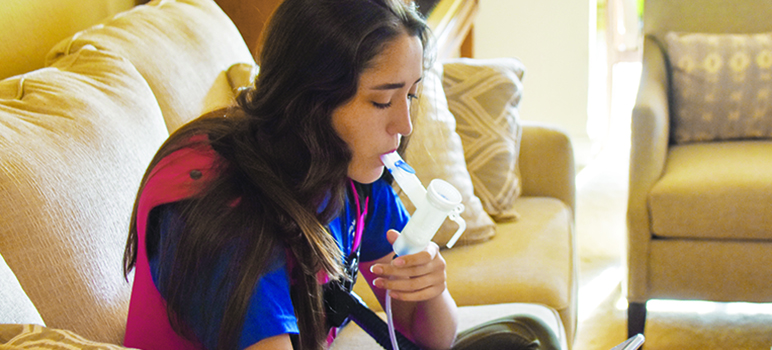
On the morning of Oct. 21, Lou Ann Alexander opened her email to find the news she’d been waiting nearly 16 years to read. “I was completely stunned,” she recalls. “I’m hoping this is what we’ve been praying for.”
When Alexander gave birth to Claire in June 2003, she seemed like a healthy infant. Then, at six months old, she developed pneumonia. Realizing that her oxygen level fell dangerously low, her pediatrician immediately admitted her to the hospital, where she spent a harrowing week. In the months that followed, Claire kept getting sick until finally, at 15 months old, a blood test revealed that she had a rare genetic disease: cystic fibrosis.
When Alexander, an optometrist in Santa Clara, looked up cystic fibrosis online, she learned that Claire’s body was producing abnormally thick, sticky mucus. It coated her lungs and obstructed her pancreas, causing frequent pulmonary infections and digestive issues. When Alexander read that the progressive disease would likely take Claire’s life in her early thirties, she says she “fell apart.”
A decade-and-a-half later, that bleak prognosis has changed drastically. On Oct. 21, the US Food and Drug Administration (FDA) approved a revolutionary new drug called Trikafta. Now, 90 percent of cystic fibrosis patients—including Claire—have a treatment that targets the underlying cause of the disease.
It has been a long road to this point. In June 1989, Dr. Lap-Chee Tsui and Dr. Francis Collins, two geneticists, pinpointed the gene responsible for cystic fibrosis, the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The two scientists toasted their discovery with coffee mugs of Canadian whiskey.
It was “a moment of celebration, but also a realization: now we have the real work to do,” said Collins, speaking to the annual North American Cystic Fibrosis Conference in Nashville, Tennessee, at the end of October.
In the decade that followed, scientists experimented with gene therapy, a method of replacing defective genes with healthy ones, but to no avail. Eventually, in 2000, the Cystic Fibrosis Foundation—a non-profit started in 1955 by a group of parents who didn’t expect their children to make it to kindergarten—invested $40 million in a biotech firm in San Diego, California called Aurora BioSciences. Aurora had developed technology that could rapidly test chemical compounds on cells. The foundation wanted the company to train its sites on cystic fibrosis drugs.
The concept of a charity giving millions of dollars to a for-profit company—known as venture philanthropy—was unorthodox, to say the least. “To go from the basic research—the academic research—to actually starting to develop new therapies, there needs to be a partnership, oftentimes with companies,” Dr. Michael Boyle, the senior vice president of therapeutics development at the foundation, explains. “They’re the ones who actually develop new drugs, not universities.”
Cystic fibrosis is rare, and there was no research suggesting that the root cause of the disease’s symptoms—dysfunctional CFTR protein within cells—could be fixed with a drug. Initially, pharmaceutical companies weren’t willing to gamble their R&D budgets on it, so the foundation stepped in and funded the early research, eliminating the financial risk. Unlike venture capitalism, the high risk-high reward system where investors fund start-ups hoping for a big payoff, the “big payoff” the foundation bet on was a lifesaving drug therapy.
In 2001, Vertex Pharmaceuticals bought Aurora for its rapid drug screening technology. The company had a choice to make: keep researching cystic fibrosis, or focus on more prevalent diseases? If it hadn’t been for the funding from the foundation, Vertex would have discontinued the cystic fibrosis program. Although the foundation’s investment was small in the world of big pharma—the foundation has invested a total of $150 million in Vertex’s R&D program, Vertex has invested over $6 billion—the timing proved crucial.
At first, when Vertex scientists set out to develop a drug to treat cystic fibrosis by fixing a tiny defective protein—a rare feat in science—others in the pharmaceutical industry were dubious. “People actually laughed,” Vertex CEO Dr. Jeff Leiden recalls. “Seriously. It was just like, there’s no way in the world that could be done.”
That didn’t deter the team at Vertex. The researchers pressed on through brute force, eventually testing not a few targeted chemical compounds, but well over a million.
Before they even got to the point of testing potential drugs, however, a litany of obstacles stood in their way. First, they had to understand the biology of the disease. Next, figure out how to clone lung cells from cystic fibrosis patients. Then, develop a method to see whether any of the chemical compounds worked on the test cells.
Finally, after they’d identified a few promising candidates, work out how to get the medicine into a pill and design clinical trials.
“It takes hundreds, sometimes thousands, of people working for decades to get a drug like this—a breakthrough drug—discovered, developed, and approved,” Leiden says.
As Vertex rushed to produce new drugs, patients like Claire worked to stay healthy. They were racing the clock. As cystic fibrosis patients get older, their lung function typically deteriorates due to infections, inflammation, and obstructions caused by the thick mucus. “What we want to do is maintain her current lung function for as long as we can,” Dr. Michael Tracy, Claire’s cystic fibrosis doctor and a pediatric pulmonologist at Stanford Children’s Health, explains.
Claire, who doesn’t think of herself as a sick kid, has another motivation for keeping her lung function high. She loves to dance. As Claire practiced ballet combinations at The New Ballet School in San Jose on a recent Saturday morning, she moved from plié to pirouette to arabesque, every muscle of her body held in perfect tension.
“Technically, she’s very strong, and she has gorgeous artistry as well,” remarks Sarah Stevens, one of her instructors.

Thanks to a groundbreaking new drug, Claire Alexander will be able to keep dancing for hours a day at The New Ballet School in San Jose. (Photo by Anne Daugherty)
Claire spent July as a student with the American Ballet Theatre’s highly selective summer intensive program in New York City. Now, she spends two-and-a-half hours every day rehearsing for The New Ballet School’s upcoming production of The Nutcracker.
Staying healthy enough to keep dancing takes work. Twice a day Claire dons a high frequency chest wall oscillation vest, which shakes her lungs and dislodges the mucus. She uses inhalers and a nebulizer to take medications that open her airways, thin out the mucus, and then irritate her lungs, forcing her to cough up phlegm.
Strict adherence to the treatment plan is key. “You never get a vacation from CF [cystic fibrosis],” Alexander says. “Ever.”
Claire also takes 30 pills a day, including three to four digestive enzyme pills whenever she eats. “She likes to boast about how many pills she can take at once,” her father Blake Alexander jokes. Her record is 12.
Claire, a reserved teenager with a droll sense of humor, tries to stay positive. She reveals the one good part of having cystic fibrosis. “I get to skip the lines at Disneyland,” she says with a shrug and a smile.
Ever since Claire’s diagnosis, doctors told her mom, dad, and older brother to keep her healthy because promising drugs lay just around the corner. The doctors hoped those drugs would be available by the time Claire turned 6, but that birthday came and went.
Finally, in 2012, there flashed a glimmer of hope. The FDA approved Vertex’s first cystic fibrosis drug, Kalydeco. The drug only treated about 4 percent of cystic fibrosis patients—there are over 1,700 genetic mutations, each of which affects protein formation in a different way—but proved it possible to treat the underlying cause of the disease. Claire’s family was hopeful. “Even if it’s not for your kid, you’re happy,” Lou Ann Alexander says.
Kalydeco was a big success for Vertex, and a proof of concept for the drugs that would follow. Leiden explains that although Vertex didn’t yet have the medicines to treat the majority of patients, “we felt we understood the biology in a way that could lead us there.” The Cystic Fibrosis Foundation sold its royalty rights for $3.3 billion, directing that money back into research.
Over the next six years, Vertex came out with its second and third cystic fibrosis drugs: Orkambi and Symdeko, which could treat about 50 percent of patients. Unfortunately, none of the drugs worked for Claire. Her health started to slip. In March, Claire spent 10 days in the hospital with a nasty bout of bronchitis that plunged her lung function from the high 90s down to 83 percent.
Then, on Oct. 21, the trajectory of Claire’s life changed. When Alexander first heard the news, she was hesitant. “You wonder sometimes if it’s too good to be true,” she says. She and her family had been waiting, hoping, praying for nearly 16 years. She started reading about the clinical trials.
In results published in The New England Journal of Medicine on Oct. 31, researchers found that after taking Trikafta for a period of six months, cystic fibrosis patients similar to Claire improved by 13.8 percent on pulmonary function tests, which measure how much air a patient can expel over one second. The average doesn’t tell the whole story, though. Some patients saw improvements of over 25 percent. “Amazing! Isn’t that amazing?” Alexander exclaims.
Perhaps even more exciting, patients in the trial experienced an average of 63 percent fewer lung infections. When it came to infections that led to hospitalizations or that required intravenous antibiotics, that drop was even higher. Those numbers mean Claire will spend fewer days in the hospital and more days dancing ballet.
Alexander struggles to put her emotions into words.
“I felt like the entire time we’ve been battling time,” she says about waiting for something revolutionary to come along. “I think it’s finally here.”
Claire, too, is excited. She read a testimonial by Gunner Esiason, a 28-year-old cystic fibrosis advocate and one of the patients in the trial. “Could I actually tell that it was working right away?” he wrote in a blog post. “You bet your ass I could.”
“I’m excited to see if it works like that,” Claire says when she first heard the news. “I hope it really works.”
Claire began taking Trikafta on Nov. 16, as soon as her insurer approved it. The drug has a hefty price tag—$311,503 per year—but with coverage and a Vertex patient support program, the Alexanders pay $15 per month. Since Claire started Trikafta she’s been coughing less, her heavy cough has all but disappeared, and she feels more energetic.
Although Trikafta is life-changing, 10 percent of patients still lack a therapy that treats the underlying cause of their disease. On Oct. 30, the foundation announced that it would channel $500 million into research on new cystic fibrosis therapies through 2025.
The cystic fibrosis community did take a moment to celebrate the milestone, though. At the annual North American Cystic Fibrosis Conference the last week of October, Collins, one of the researchers who discovered the gene back in 1989, took the stage.
Every time there is a breakthrough in cystic fibrosis research, he performs his original song, “Dare to Dream.” This year, wearing a DNA double helix-patterned tie and strumming a black acoustic guitar, Collins added a new verse.
“To find that triple treatment has taken 30 years, and it took the skills and vision of a team,” he sang. “There were times when nothing seemed to work, and progress seemed to slow. But hope is in our genes, we dare to dream.”
The crowd rose in a standing ovation.
My 24 year old son is one of the people in the 90% of cystic fibrosis patients who can actually benefit from this drug, thank goodness. However, we are now facing an appeal after waiting two weeks for the clinic to submit the information to our insurance, and then having the authorization denied by the insurance. Crossing fingers that my son gets to try Trikafta — and trying to keep him as healthy as possible in the meantime.